ņä£ļĪĀ
ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æ(Alzheimer disease)ņØĆ ņ╣śļ¦żņØś Ļ░Ćņן ņŻ╝ņÜöĒĢ£ ņøÉņØĖ ņ¦łĒÖśņØ┤ļ®░, ņĄ£ĻĘ╝ņØś ņØĖĻĄ¼ Ļ│ĀļĀ╣ĒÖö ņČöņäĖņÖĆ ĒĢ©Ļ╗ś ĻĖēĻ▓®Ē׳ ņ”ØĻ░ĆļÉśĻ│Ā ņ׳ļŗż[1]. ņØ┤ļ¤¼ĒĢ£ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņØś ņ╣śļŻīņŚÉ ņ׳ņ¢┤ņä£ ļ░£ļ│æ ĻĖ░ņĀäņ£╝ļĪ£ ņé¼ņØ┤ĒåĀņ╣┤ņØĖ(cytokine) ļ¦żĻ░£ ņŗĀĻ▓ĮņŚ╝ņ”Øļ░śņØæņŚÉ ņ┤łņĀÉņØä ļ¦×ņČś ņŚ░ĻĄ¼ļōżņØ┤ ĒÖ£ļ░£Ē׳ ņØ┤ļżäņ¦ĆĻ│Ā ņ׳ļŖöļŹ░ ĻĘĖ ņżæņŚÉņä£ ņóģņ¢æĻ┤┤ņé¼ņØĖņ×É-╬▒ (tumor necrosis factor [TNF]-╬▒)Ļ░Ć ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż[2]. ņĢīņĖĀĒĢśņØ┤ļ©Ė ņźÉ ļ¬©ļŹĖ ņŚ░ĻĄ¼ņŚÉņä£ TNF-╬▒Ļ░Ć ├¤-secretaseņÖĆ ╬│-secretase ļ░£ĒśäņØä ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ņä£ ╬▓-ņĢäļ░ĆļĪ£ņØ┤ļō£(╬▓-amyloid, A╬▓)ņØś ņāØņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āĻ│╝ TNF-╬▒ņŚÉ ņśżļל ļģĖņČ£ļÉśļŖö Ļ▓ĮņÜ░ ņŗĀĻ▓ĮņäĖĒżņØś Ļ┤┤ņé¼Ļ░Ć ņ┤łļלļÉ£ļŗżļŖö Ļ▓ā ļō▒ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņŚÉņä£ TNF-╬▒ņØś ļ│æļ”¼ĻĖ░ņĀäņāü ņŚ░Ļ┤Ćņä▒ņØ┤ ļ│┤Ļ│ĀļÉ£ ļ░ö ņ׳ļŗż[3,4]. ļśÉ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æ ĒÖśņ×ÉņØś ĒśłņĢĪ ļé┤ TNF-╬▒ ņłśņ╣śĻ░Ć ņĀĢņāüņØĖņŚÉ ļ╣äĒĢ┤ ņ£ĀņØśĒĢśĻ▓ī ļåÆņĢśĻ│Ā, ļćīņ▓ÖņłśņĢĪņŚÉņä£ļÅä ļŗżļźĖ ņé¼ņØ┤ĒåĀņ╣┤ņØĖ(TNF-╬▓, interleukin [IL]-1╬▓, IL-6)Ļ│╝ ļŗ¼ļ”¼ TNF-╬▒ņØś ņłśņ╣śĻ░Ć ņ”ØĻ░ĆļÉ£ Ļ▓āņØ┤ ĒÖĢņØĖļÉśņŚłļŗż[5,6]. ĻĘĖļ¤¼ļéś ņĢäņ¦ü TNF-╬▒ ņ¢ĄņĀ£ņĀ£ļź╝ ņØ┤ņÜ®ĒĢ£ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņØś ņŚ░ĻĄ¼ļŖö ĻĘĖ ņłśĻ░Ć ļ¦żņÜ░ ņĀüņØä ļ┐É ņĢäļŗłļØ╝ etanercept, infliximab ļō▒ņØä ņé¼ņÜ®ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ Ļ▓░Ļ│╝Ļ░Ć ņØ╝ņĀĢĒĢśņ¦Ć ļ¬╗ĒĢśņśĆļŗż[2].
ņØ┤ņŚÉ ļ│Ė ņŚ░ĻĄ¼ņ¦äņØĆ ņØ┤ļ»Ė ļźśļ¦łĒŗ░ņŖż Ļ┤ĆņĀłņŚ╝ ņ¦łĒÖśņŚÉņä£ ļäÉļ”¼ ņé¼ņÜ®ļÉśļŖö ņŻ╝ņÜö TNF-╬▒ ņ¢ĄņĀ£ņĀ£ ņżæ ĒĢśļéśņØĖ adalimumabņØä ņé¼ņÜ®ĒĢśņŚ¼ ņĢīņĖĀĒĢśņØ┤ļ©Ė ĒĢ┤ļ¦ł ņĀłĒÄĖ ļ¬©ļŹĖņŚÉņä£ņØś ĒÜ©Ļ│╝ļź╝ ĒÖĢņØĖĒĢ┤ ļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
ļ░®ļ▓Ģ
1. ņŗżĒŚś ņźÉ ĒĢ┤ļ¦łņØś ĻĖ░Ļ┤ĆĒśĢņĀü ņĀłĒÄĖļ░░ņ¢æ
ņŗżĒŚś ņźÉ ĒĢ┤ļ¦ł ļ░░ņ¢æņØĆ Stoppini ļō▒[7]ņŚÉ ņØśĒĢ┤ ĻĖ░ņłĀļÉ£ ļ░®ļ▓ĢņØä ĒåĀļīĆļĪ£ ņ¦äĒ¢ēĒĢśņśĆļŗż. ņāØĒøä 6ņØ╝ļÉ£ ņŗżĒŚś ņźÉ(Sprague-Dawley rats)ļź╝ Ńł£ņśżļ”¼ņŚöĒŖĖļ░öņØ┤ņśżļĪ£ļČĆĒä░ ĻĄ¼ņ×ģĒĢśņŚ¼ 1ņØ╝Ļ░ä Ļ▓ĆņŚŁ ĻĖ░Ļ░äņØä ļæö Ēøä, 7ņØ╝ļĀ╣ ņŗżĒŚś ņźÉļĪ£ļČĆĒä░ ĒĢ┤ļ¦łņØś ĻĖ░Ļ┤ĆĒśĢņĀü ņĀłĒÄĖļ░░ņ¢æņØä ņŗ£Ē¢ēĒĢśņśĆļŗż. ļæÉĒö╝ļź╝ 70% ņĢīņĮöņś¼ļĪ£ ņåīļÅģĒĢ£ Ēøä ļ¼┤ĻĘĀņĀüņ£╝ļĪ£ ņĀä ļćīļź╝ ļ░Ģļ”¼ĒĢśĻ│Ā 4Ōäā dissection mediaĻ░Ć ļŗ┤ĻĖ┤ ĒÄśĒŖĖļ”¼ņĀæņŗ£ļĪ£ ņś«Ļ▓╝ļŗż. ļŗżņØīņ£╝ļĪ£ ņ¢æņ¬Į ĒĢ┤ļ¦łļČĆņ£äļź╝ ļČäļ”¼ĒĢśņŚ¼ ņłśļÅÖ ņĪ░ņ¦ü ņĀłļŗ©ĻĖ░(manual tissue chopper)ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ 450 ┬Ąm ļæÉĻ╗śļĪ£ ņĀłĒÄĖņØä ļ¦īļōĀ Ēøä, ņØ┤ ņżæ ņåÉņāüņØ┤ ņŚåļŖö ņóŗņØĆ ņāüĒā£ņØś ņĀłĒÄĖļ¦īņØä ņäĀĒāØĒĢśņŚ¼ porous membrane insert (Micropore)ļĪ£ ļ░░ņ╣śĒĢśņśĆļŗż. InsertsļŖö 6 well platesņŚÉ ņ£äņ╣śņŗ£ĒéżĻ│Ā 1Ļ░£ņØś insertņŚÉļŖö 5-6Ļ░£ņØś ņĀłĒÄĖņØä ļ░░ņŚ┤ĒĢśņśĆļŗż. ņĪ░ņ¦ü ņŻ╝ļ│ĆņØś dissection mediaļź╝ ņĀ£Ļ▒░ĒĢ£ ļÆż 1 mlņØś culture media (50% MEM, 25% HBSS, 25% heat inactivated horse serum, supplemented with D-glucose to 6.5 mg/ml and glutamine 1 mM, pH 7.3, filtered)ļĪ£ Ļ░łņĢäņŻ╝ņŚłļŗż. ļ¬©ļōĀ Ļ│╝ņĀĢņØ┤ ņÖäļŻīļÉ£ ņĀłĒÄĖņØ┤ ļōżņ¢┤ņ׳ļŖö 6 well platesļź╝ 36ŌäāņØś humidified 5% CO2 incubatorļĪ£ ņś«ĻĖ┤ Ēøä 3ņØ╝ Ļ░äĻ▓®ņ£╝ļĪ£ ļ░░ņ¢æņĢĪņØä Ļ░łņĢäņŻ╝ļ®░ ĒĢ┤ļ¦łņĪ░ņ¦üņØä ņ¦ĆņåŹņĀüņ£╝ļĪ£ ļ░░ņ¢æĒĢśņśĆļŗż. ĻĖ░ņłĀļÉ£ ņĀä Ļ│╝ņĀĢņØĆ horizontal laminar flow hoodņŚÉņä£ ņŚäĻ▓®ĒĢ£ ļ¼┤ĻĘĀ ĻĖ░ļ▓Ģņ£╝ļĪ£ ņ¦äĒ¢ēļÉśņŚłļŗż.
2. A╬▓ ļ░Å adalimumab ņ▓śļ”¼
A╬▓1-42 (#62-0-80B; American Peptide)ļŖö ņé¼ņÜ® ņ¦üņĀäņŚÉ 37ŌäāņŚÉņä£ 24ņŗ£Ļ░ä ļÅÖņĢł ļ░░ņ¢æĒĢśņŚ¼ oligomerļź╝ ņĀ£ņ×æĒĢśņśĆļŗż. ņŗżĒŚśĻĄ░ņØĆ ļīĆņĪ░ĻĄ░, A╬▓1-42 ļŗ©ļÅģ ņŗżĒŚśĻĄ░, A╬▓1-42ņÖĆ adalimumab (Humira; Eisai Korea Inc.) ļÅÖņŗ£ ņŗżĒŚśĻĄ░ņØś ņäĖ ĻĄ░ņ£╝ļĪ£ ļéśļłäņ¢┤ Ļ░ü ĻĄ░ņŚÉ 2 well platesņö®ņØä ļ¼┤ņ×æņ£äļĪ£ ļ░░ņĀĢĒĢśņśĆļŗż. ļ░░ņ¢æ 14ņØ╝ņ¦ĖņŚÉ A╬▓1-42 ņŗżĒŚśĻĄ░Ļ│╝ A╬▓1-42ņÖĆ Adalimumab (Humira) ļÅÖņŗ£ ņŗżĒŚśĻĄ░ņØä ĻĄ¼ļČäĒĢśņŚ¼ ņŗżĒŚś ļīĆņāüņØ┤ ļÉĀ insertņŚÉ A╬▓1-42ņÖĆ adalimumabļź╝ ņĀüņĀĢ ļåŹļÅäļĪ£ ņĀÉņĀüĒĢ┤ņŻ╝ņŚłļŗż. ĒĢ┤ļ¦łņĪ░ņ¦üņŚÉ 100 ┬ĄM oligomer A╬▓1-42ļź╝ ņØ┤ĒŗĆņŚÉ ĒĢ£ ļ▓ł Ļ░äĻ▓®ņ£╝ļĪ£ ņĀÉņĀüĒĢśņśĆņ£╝ļ®░, adalimumabļŖö 100 ┬Ąg/ml ļåŹļÅäļĪ£ 3ņØ╝ļ¦łļŗż ĒĢ£ ļ▓łņö® ņĀÉņĀüĒĢśņśĆļŗż[8-10]. ņØ┤ Ēøä, 9ņØ╝ ļÅÖņĢł 36Ōäā humidified 5% CO2 incubatorņŚÉņä£ ļ░░ņ¢æĒĢśļ®┤ņä£ 3ņØ╝ņŚÉ ĒĢ£ ļ▓łņö® ļ░░ņ¢æņĢĪņØä ņāłļĪŁĻ▓ī ĻĄÉņ▓┤ĒĢ┤ņŻ╝ņŚłļŗż.
3. Propidium iodideļź╝ ņØ┤ņÜ®ĒĢ£ ņŗĀĻ▓ĮņøÉ ņåÉņāüņØś ĒÅēĻ░Ć
ļ░░ņ¢æ 14ņØ╝ņ¦Ė Ļ░üĻ░üņØś insertņŚÉ ĒżĒĢ©ļÉśņ¢┤ņ׳ļŖö ļ░░ņ¢æņĢĪņØä ļ¬©ļæÉ ņĀ£Ļ▒░ĒĢ£ Ēøä propidium iodide (PI, 1 ┬Ąg/ml in serum free media; Sigma)ļź╝ ņ▓©Ļ░ĆĒĢśņśĆļŗż. 24ņŗ£Ļ░ä ļÅÖņĢł ļ░░ņ¢æĒĢ£ ļŗżņØī ņŚŁĒśĢĻ┤æ Ēśäļ»ĖĻ▓Į(inverted fluorescence microscope)ņØä ņØ┤ņÜ®ĒĢśņŚ¼ ĒĢ┤ļ¦łņĪ░ņ¦üņØś ĻĄ¼ņĪ░ņÖĆ ņŗĀĻ▓ĮņäĖĒż ļČäĒż ņāüĒā£ ļ░Å ĒśĢĻ┤æļ░£Ēśä ņ¢æņāüņØä Ļ┤Ćņ░░ĒĢśņśĆļŗż.
4. Western blotņØä ņØ┤ņÜ®ĒĢ£ ļŗ©ļ░▒ņ¦ł ļ░£ĒśäļČäņäØ
72ņŗ£Ļ░ä ļÅÖņĢł 36Ōäā CO2 incubatorņŚÉņä£ ļ░░ņ¢æĒĢ£ ĒĢ┤ļ¦łņĪ░ņ¦üļōżņØä Ļ░ü ĻĄ░ļ│äļĪ£ ļ¬©ņĢä Protease Inhibitor Cocktail (Sigma)Ļ│╝ Phosphatase Inhibitor Cocktail 1 (Sigma)ņØä ņ▓©Ļ░ĆĒĢ£ RIPA buffer (Sigma)ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ļŗ©ļ░▒ņ¦łņØä ņČöņČ£ĒĢśņśĆļŗż. ņżĆļ╣äļÉ£ ļŗ©ļ░▒ņ¦ł ņŗ£ļŻīļŖö Pierce BCA Protein Assay Kit (Thermo)ļź╝ ņé¼ņÜ®ĒĢ┤ ņĀĢļ¤ēĒĢ£ ļÆż, 30 ┬ĄgļĪ£ Ļ░ü ņŗżĒŚśĻĄ░ņŚÉņä£ ļÅÖņØ╝ĒĢ£ ļåŹļÅäņØś ļŗ©ļ░▒ņ¦łņØä loadingĒĢśņŚ¼ SDS-PAGEļź╝ ņŗżņŗ£ĒĢśņśĆļŗż. ļŗżņØīņ£╝ļĪ£ transfer bufferņŚÉņä£ nitrocellulose membrane (Biorad)ņ£╝ļĪ£ ļŗ©ļ░▒ņ¦łļōżņØä ņØ┤ļÅÖņŗ£ņ╝£ western blotņØä ņŗżņŗ£ĒĢśņśĆļŗż. 0.1 % ponceau SļĪ£ ņŚ╝ņāēĒĢśņŚ¼ ļŗ©ļ░▒ņ¦łņØś ņĀäĻĖ░ņśüļÅÖ ņāüĒā£ļź╝ Ļ┤Ćņ░░ĒĢśņśĆĻ│Ā, TBSTļĪ£ ņŚ¼ļ¤¼ ļ▓ł ņö╗ņ¢┤ ponceau Sļź╝ ņĀ£Ļ▒░ĒĢśņśĆļŗż. MembraneņØä 5% nonfat dry skim milk ļśÉļŖö 5% BSAĻ░Ć ĒżĒĢ©ļÉ£ TBSTņØś blocking solutionņŚÉ ļäŻņ¢┤ ņāüņś©ņŚÉņä£ 1ņŗ£Ļ░ä ļÅÖņĢł ļ░śņØæņŗ£ņ╝░ļŗż. Primary antibodyļŖö blocking solutionņŚÉ 1:500ņ£╝ļĪ£ ĒؼņäØņŗ£ņ╝£ 4ŌäāņŚÉņä£ ļ░ż ņé¼ņØ┤ņŚÉ ļ░śņØæņŗ£Ēé© Ēøä, TBSTļĪ£ 10ļČäņö® ņäĖ ļ▓ł ņäĖņ▓ÖĒĢśņśĆĻ│Ā, 1:1,000ņ£╝ļĪ£ ĒؼņäØĒĢ£ HRP-conjugated secondary antibodyņÖĆ ņāüņś©ņŚÉņä£ 1ņŗ£Ļ░ä ļÅÖņĢł ļ░śņØæņŗ£ņ╝░ļŗż. TBSTļĪ£ 10ļČäņö® 3ļ▓ł ņäĖņ▓ÖĒĢ£ ļÆż ECL Start Western Blotting Detection Reagent (Amersham)ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ļŗ©ļ░▒ņ¦ł ļ░£ĒśäņØä Ļ░Ćņŗ£ĒÖöĒĢśņśĆļŗż. ņé¼ņÜ®ļÉ£ primary antibodyļĪ£ļŖö NeuN (#12943; Cell Signaling), Bcl2 (#2870; Cell Signaling), ╬▓-actin (#3700; Cell Signaling), pTau181 (#12885; Cell Signaling), Tau (#4019; Cell Signaling)ļź╝ ņé¼ņÜ®ĒĢśņśĆļŗż. Secondary antibodyļĪ£ļŖö bovine anti-rabbit IgG-HRP (sc-2370; Santa CruZ Biotechnology), goat anti-mouse IgG-HRP (sc-2005; Santa CruZ Biotechnology)ļź╝ ņé¼ņÜ®ĒĢśņśĆļŗż. Western blot ļ░┤ļō£ļŖö ImageJ 1.49 ProgramņØä ņØ┤ņÜ®ĒĢśņŚ¼ ļåŹļÅäļź╝ ĻĄ¼ĒĢ©ņ£╝ļĪ£ņä£ Ļ░ü ļŗ©ļ░▒ņ¦łņØś ļ░£Ēśä ņ¢æņØä ļ╣äĻĄÉļČäņäØĒĢśņśĆļŗż.
Ļ▓░Ļ│╝
1. AdalimumabņØś ņŗĀĻ▓ĮņäĖĒż ļ│┤ĒśĖ ĒÜ©Ļ│╝
100 ┬ĄM A╬▓ ņ▓śļ”¼ĻĄ░Ļ│╝ 100 ┬ĄM A╬▓1-42ņÖĆ 100 ┬Ąg/ml adalimumabņØä ļÅÖņŗ£ņŚÉ ņ▓śļ”¼ĒĢ£ ĻĄ░ņŚÉņä£ ņŗĀĻ▓ĮņäĖĒż ļ│┤ĒśĖ ĒÜ©Ļ│╝ļź╝ ļČäņäØĒĢśņśĆļŗż. ĻĖ░Ļ┤ĆĒśĢņĀü ĒĢ┤ļ¦łņĪ░ņ¦ü ņĀłĒÄĖļ░░ņ¢æ ĻĖ░ņłĀņØä ĒåĄĒĢśņŚ¼ ņźÉ ĒĢ┤ļ¦łņĪ░ņ¦üņØä 6 well platesņØś insert ņ£äņŚÉ ņ£äņ╣śņŗ£Ēé© Ēøä, CO2 incubatorņŚÉņä£ ļ░░ņ¢æņØä ņŗżņŗ£ĒĢśņśĆļŗż. ļ░░ņ¢æ 14ņØ╝ņ¦Ė ļÉśļŖö ļéĀļČĆĒä░ A╬▓1-42ņÖĆ adalimumabņØä ņØ╝ņĀĢ ļåŹļÅäļĪ£ 2ņØ╝, 3ņØ╝ Ļ░äĻ▓®ņ£╝ļĪ£ ņĀÉņĀüĒĢ£ ļÆż PI ņŚ╝ņāēņØä ņŗżņŗ£ĒĢśņśĆļŗż(Fig. 1). ļīĆņĪ░ĻĄ░Ļ│╝ ļ╣äĻĄÉĒĢśņśĆņØä ļĢī 100 ┬ĄM A╬▓1-42 ņŗżĒŚśĻĄ░ņŚÉņä£ ĒĢ┤ļ¦ł ņĀłĒÄĖ CA1 ļČĆļČäņØś ņåÉņāüņØä ļ│┤ņŚ¼ņŻ╝ļŖö PI-positiveĒĢ£ ĒśĢĻ┤æļ░£Ēśä ļ®┤ņĀüņØ┤ ņ£ĀņØśĒĢśĻ▓ī ņ”ØĻ░ĆĒĢśņśĆļŗż(p=0.018). TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņØĖ adalimumabņØä 100 ┬ĄM A╬▓1-42ņÖĆ ĒĢ©Ļ╗ś 100 ┬Ąg/mlņØś ļåŹļÅäļĪ£ ņĀÉņĀüĒĢ£ ĻĄ░ņŚÉņä£ļŖö PI ĒśĢĻ┤æļ░£ĒśäņØ┤ A╬▓1-42ļ¦īņØä ņ▓©Ļ░ĆĒĢ£ ņŗżĒŚśĻĄ░ļ│┤ļŗż ņ¢ĄņĀ£ļÉśņ¢┤ ņäĖĒżņé¼ļ®ĖņØ┤ Ļ░ÉņåīļÉ£ Ļ▓āņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳ņŚłļŗż(p=0.035). ņØ┤ļź╝ ĒåĄĒĢ┤ adalimumabņØ┤ A╬▓1-42ļĪ£ ņØĖĒĢ£ ņäĖĒżņé¼ļ®ĖņØä ļ¦ēņĢäņŻ╝ļŖö ņŗĀĻ▓ĮņäĖĒż ļ│┤ĒśĖ ĒÜ©ļŖźņØä Ļ░Ćņ¦ĆļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż.
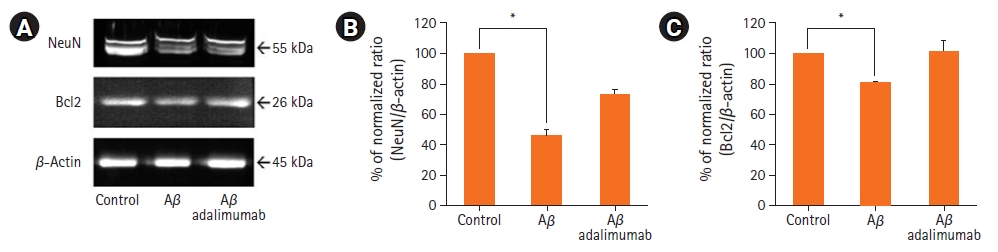
2. NeuNĻ│╝ Bcl2 ļŗ©ļ░▒ ļ░£ĒśäņØś ņ¢ĄņĀ£
A╬▓1-42ļĪ£ ņØĖĒĢ£ ļÅģņä▒ņŚÉ ļīĆĒĢ£ adalimumabņØś ņŗĀĻ▓Įļ│┤ĒśĖ ĒÜ©Ļ│╝ļź╝ ĒÖĢņØĖĒĢśĻĖ░ ņ£äĒĢśņŚ¼ Ļ░ü ĻĄ░ņØś ĒĢ┤ļ¦łņĪ░ņ¦ü ņĀłĒÄĖņ£╝ļĪ£ļČĆĒä░ ļŗ©ļ░▒ņ¦łņØä ņČöņČ£ĒĢśņŚ¼ western blotņØä ņłśĒ¢ēĒĢśņśĆļŗż(Fig. 2). ņŗĀĻ▓ĮņøÉ ņäĖĒżņØś Ēæ£ņŗØņ×ÉņØĖ NeuNĻ│╝ ĒĢŁ-ņäĖĒżņé¼ļ®Ė(anti-apoptosis) ļŗ©ļ░▒ņØ┤ļ®┤ņä£ ņĪ░ņ¦üņØś ļ░£ļŗ¼Ļ│╝ ļČäĒÖöņŚÉ Ļ┤ĆņŚ¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņ¦ä Bcl2 ļŗ©ļ░▒ņ¦łņØś ļ░£ĒśäņØä ņĖĪņĀĢĒĢśņśĆļŗż. ļīĆņĪ░ĻĄ░Ļ│╝ ļ╣äĻĄÉĒĢśņśĆņØä ļĢī NeuNĻ│╝ Bcl2 ļŗ©ļ░▒ņ¦łņØś ļ░£ĒśäņØĆ 100 ┬ĄMņØś A╬▓1-42ņŚÉ ņØśĒĢ┤ ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīĒĢśņśĆļŗż(p=0.04, p=0.02). 100 ┬Ąg/ml adalimumabĻ│╝ A╬▓1-42Ļ░Ć ĒĢ©Ļ╗ś ņ▓śļ”¼ļÉ£ ĻĘĖļŻ╣ņŚÉņä£ļŖö adalimumabņØ┤ ĒżĒĢ©ļÉśņ¦Ć ņĢŖņØĆ ļ░░ņ¦ĆņŚÉņä£ ļ░░ņ¢æĒĢ£ ņĪ░ņ¦üĻĄ░ļ│┤ļŗż NeuNņÖĆ Bcl2 ļŗ©ļ░▒ņ¦ł ļ░£Ēśä ņ¢æņØ┤ ņ£ĀņØśĒĢśņ¦Ć ņĢŖņĢśņ£╝ļéś ņ”ØĻ░ĆĒĢśļŖö Ļ▓ĮĒ¢źņØä ļ│┤ņśĆĻ│Ā, ļīĆņĪ░ĻĄ░Ļ│╝ ļ╣äĻĄÉĒĢśņśĆņØä ļĢī ņ£ĀņØśĒĢ£ Ļ░ÉņåīļÅä Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśļŗż.
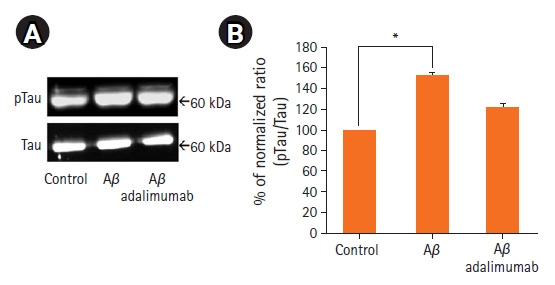
3. Tau ļŗ©ļ░▒ Ļ│╝ņØĖņé░ĒÖöņØś ņ¢ĄņĀ£
ņźÉ ĒĢ┤ļ¦ł ņĀłĒÄĖņŚÉņä£ A╬▓1-42ļĪ£ ņØĖĒĢ£ ņŗĀĻ▓ĮļÅģņä▒ņØ┤ tau ļŗ©ļ░▒ņ¦łņØś Ļ│╝ņØĖņé░ĒÖöņÖĆ Ļ┤ĆļĀ© ņ׳ļŖöņ¦Ć ņĢīņĢäļ│┤ĻĖ░ ņ£äĒĢśņŚ¼ Ļ│╝ņØĖņé░ĒÖöļÉ£ tau ļŗ©ļ░▒ņ¦łņØś ļ░£Ēśä ņĀĢļÅäļź╝ western blotņ£╝ļĪ£ ĒÖĢņØĖĒĢśņśĆļŗż(Fig. 3). 100 ┬ĄMņØś A╬▓1-42 ņ▓śļ”¼ĻĄ░ņŚÉņä£ Ļ│╝ņØĖņé░ĒÖöļÉ£ tauņØś ļŗ©ļ░▒ņ¦ł ļ░£ĒśäņĀĢļÅäĻ░Ć ļīĆņĪ░ĻĄ░ņŚÉ ļ╣äĒĢ┤ ņ£ĀņØśĒĢśĻ▓ī ņ”ØĻ░ĆĒĢśņśĆĻ│Ā(p=0.03), 100 ┬Ąg/ml adalimumabņØä ļÅÖņŗ£ņŚÉ ņ▓śļ”¼ĒĢ£ ĻĄ░ņŚÉņä£ļŖö A╬▓1-42ļ¦īņØä ņ▓©Ļ░ĆĒĢ£ ņŗżĒŚśĻĄ░ļ│┤ļŗż ņ£ĀņØśĒĢśņ¦Ć ņĢŖņĢśņ£╝ļéś Ļ░ÉņåīĒĢśļŖö Ļ▓ĮĒ¢źņØä ļ│┤ņśĆņ£╝ļ®░ ļīĆņĪ░ĻĄ░Ļ│╝ ļ╣äĻĄÉĒĢśņŚ¼ ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśļŗż. ļö░ļØ╝ņä£ A╬▓1-42ļĪ£ ņØĖĒĢ£ ņŗĀĻ▓ĮļÅģņä▒ņØĆ ņĢīņĖĀĒĢśņØ┤ļ©Ė ņ¦łļ│æņŚÉ ĒŖ╣ņØ┤ņĀüņØĖ ļŗ©ļ░▒ņ¦łņØĖ, tauņØś Ļ│╝ņØĖņé░ĒÖöļź╝ ņ”ØĻ░Ćņŗ£Ēéżļ®░ ņØ┤ļ¤¼ĒĢ£ ĒśäņāüņØĆ adalimumabļź╝ ĒĢ©Ļ╗ś ņ▓śļ”¼ĒĢśņśĆņØä Ļ▓ĮņÜ░ ņ¢ĄņĀ£ļÉśļŖö Ļ▓░Ļ│╝Ļ░Ć ĒÖĢņØĖļÉśņŚłļŗż.
Ļ│Āņ░░
ļ│Ė ņŚ░ĻĄ¼ņŚÉņä£ A╬▓1-42 ņŗżĒŚśĻĄ░ņØĆ ņŗĀĻ▓ĮņøÉ ņäĖĒżņé¼ļ®ĖļĪ£ ņØĖĒĢ┤ ĒĢ┤ļ¦łņĪ░ņ¦üņØś CA1 ļČĆļČäņØś PI uptakeĻ░Ć ņ”ØĻ░ĆļÉśņŚłĻ│Ā, ņŗĀĻ▓ĮņøÉ ņäĖĒżņØś ĒŖ╣ņØ┤ņĀü Ēæ£ņ¦Ć ļŗ©ļ░▒ņ¦łņØĖ NeuNĻ│╝ ĒĢŁ-ņäĖĒżņé¼ļ®Ė ļŗ©ļ░▒ņ¦łņØĖ Bcl2ņØś ļ░£ĒśäņØ┤ Ļ░ÉņåīļÉśņŚłļŗż. ļśÉ A╬▓1-42 ņŗżĒŚśĻĄ░ņŚÉņä£ ņäĖĒżņé¼ļ®ĖĻ│╝ Ļ┤ĆļĀ©ļÉ£ Ļ│╝ņØĖņé░ĒÖöļÉ£ tau ļŗ©ļ░▒ņ¦łņØś ļ╣äņ£©ņØ┤ ņ£ĀņØśĒĢśĻ▓ī ņ”ØĻ░ĆļÉ£ Ļ▓āņØ┤ Ļ┤Ćņ░░ļÉśņŚłļŗż. ņØ┤ļ¤¼ĒĢ£ Ļ▓Ćņé¼ Ļ▓░Ļ│╝ļŖö ņĢīņĖĀĒĢśņØ┤ļ©Ė ĒĢ┤ļ¦ł ņĀłĒÄĖ ļ¬©ļŹĖņØä ņØ┤ņÜ®ĒĢśņŚ¼ A╬▓1-42 ņŗżĒŚśĻĄ░ņØś ņŗĀĻ▓ĮļÅģņä▒ņØ┤ Ļ░ØĻ┤ĆņĀüņ£╝ļĪ£ ĒÖĢņØĖļÉ£ Ļ▓āņ£╝ļĪ£ ļ│╝ ņłś ņ׳ļŗż.
ņØ┤ļ¤¼ĒĢ£ A╬▓1-42 ņŗżĒŚśĻĄ░ņØś ņŗĀĻ▓ĮļÅģņä▒ņŚÉ ņØśĒĢ£ ņŗĀĻ▓ĮņøÉ ņäĖĒż ņåÉņāü ļ░Å ņé¼ļ®ĖņØĆ A╬▓1-42ņÖĆ ĒĢ©Ļ╗ś TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņØĖ adalimumabņØä ņ▓śļ”¼ĒĢśņśĆņØä ļĢī A╬▓1-42ņŚÉ ņØśĒĢ┤ ņ£ĀļÅäļÉśņŚłļŹś PI uptake ņ”ØĻ░Ć, NeuNĻ│╝ Bcl2ņØś ļ░£Ēśä Ļ░Éņåī, Ļ│╝ņØĖņé░ĒÖöļÉśņŚłļŹś tau ļŗ©ļ░▒ņ¦ł ļ╣äņ£©ņØś ņ”ØĻ░Ć ļō▒ņØ┤ ļ░£ĒśäļÉśņ¦Ć ņĢŖļŖö Ļ▓āņ£╝ļĪ£ ļ│┤ņĢä adalimumabņØ┤ A╬▓1-42ņŚÉ ņØśĒĢ£ ņŗĀĻ▓ĮļÅģņä▒ņØä ņÖäĒÖöņŗ£ņ╝£ņŻ╝ļŖö Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż.
ĻĖ░ņĪ┤ņØś ņŚ░ĻĄ¼ņŚÉņä£ TNF-╬▒ ņ¢ĄņĀ£ņĀ£ļŖö ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æ ĒÖśņ×ÉņÖĆ ļÅÖļ¼╝ļ¬©ļŹĖņŚÉņä£ ņŗĀĻ▓Į ļ│┤ĒśĖ ĒÜ©Ļ│╝ļź╝ Ļ░¢ļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż[11-13]. ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æ ņźÉņŚÉņä£ TNF-╬▒ ņ¢ĄņĀ£ņĀ£ļź╝ ļīĆļćīņŚÉ ņŻ╝ņ×ģĒ¢łņØä ļĢī ņŗĀĻ▓ĮņŚ╝ņ”ØņØś ļ¦żĻ░£ņ▓┤ņØĖ TNFļź╝ ņ¢ĄņĀ£ĒĢ©ņ£╝ļĪ£ņŹ© ĒĢ┤ļ¦ł, Ēö╝ņ¦ł, ĒÄĖļÅä ļČĆņ£äņŚÉņä£ ļ¦īņä▒ ņŚ╝ņ”ØņŚÉ ņØśĒĢ┤ ņ£Āļ░£ļÉśļŖö A╬▓ ļ│æļ”¼ ņåīĻ▓¼ņØ┤ Ļ░ÉņåīļÉ©ņØ┤ ĒÖĢņØĖļÉśņŚłļŗż[11]. TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņØĖ infliximabņØä ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æ ņźÉņØś ļćīņŗżņŚÉ ņŻ╝ņ×ģĒĢśņśĆņØä ļĢī TNF-╬▒, amyloid plaque, tau Ļ│╝ņØĖņé░ĒÖö ļō▒ņØ┤ ņżäņ¢┤ļō£ļŖö Ļ▓āņØ┤ ĒÖĢņØĖļÉśņŚłĻ│Ā TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņŚÉ ņØśĒĢ┤ ĒÖ£ņä▒ĒÖöļÉ£ CD11c positive dendritic like cellņØ┤ amyloid plaqueņØś ņĀ£Ļ▒░Ļ│╝ņĀĢņŚÉ Ļ┤ĆļĀ©ļÉ£ Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉśņŚłļŗż[12]. ņĄ£ĻĘ╝ TNFĻ░Ć ļ│æļ”¼ĻĖ░ņĀäņŚÉ Ļ┤ĆņŚ¼ļÉśĻ│Ā, ņ╣śļŻīņĀ£ļĪ£ TNF ņ¢ĄņĀ£ņĀ£Ļ░Ć ņé¼ņÜ®ļÉśļŖö ņŚ╝ņ”Øņä▒ ņ¦łĒÖśņØä Ļ░Ćņ¦ä ĒÖśņ×ÉļōżņØä ļīĆņāüņ£╝ļĪ£ ņŗżņĀ£ ņ×äņāüņ×ÉļŻī(real-world data)ļź╝ ĒÖ£ņÜ®ĒĢ£ ĒøäĒ¢źņĀü ņŚ░ĻĄ¼Ļ░Ć ņ׳ņŚłļŖöļŹ░, ļźśļ¦łĒŗ░ņŖż Ļ┤ĆņĀłņŚ╝, Ļ▒┤ņäĀ, ņŚ╝ņ”Øņä▒ ņןņ¦łĒÖś ļō▒ņØś ņŚ╝ņ”Øņä▒ ņ¦łĒÖś ĒÖśņ×ÉļōżņØĆ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņŚÉ Ļ▒Ėļ”┤ ņ£äĒŚśņØ┤ ņ£ĀņØśĒĢśĻ▓ī ļåÆņĢśņ£╝ļéś adalimumab ļō▒ TNF ņ¢ĄņĀ£ņĀ£ļź╝ ņé¼ņÜ®ĒĢ£ ĒÖśņ×ÉņØś Ļ▓ĮņÜ░ ņØ┤ļ¤¼ĒĢ£ ņ£äĒŚśņØ┤ ļé«ņĢäņ¦ä Ļ▓āņØ┤ ĒÖĢņØĖļÉ£ļ░ö ņ׳ļŗż[13].
ĒåĄņāü adalimumabĻ│╝ Ļ░ÖņØĆ TNF-╬▒ ņ¢ĄņĀ£ņĀ£ļŖö ļćīņ¦łĒÖśņŚÉ ļīĆĒĢ£ ņ╣śļŻīļź╝ ļ¬®ņĀüņ£╝ļĪ£ Ļ░£ļ░£ļÉ£ ņĢĮņĀ£Ļ░Ć ņĢäļŗłļ»ĆļĪ£ ļćīĒśłĻ┤Ć ņןļ▓Į(blood brain barrier, BBB)ņØä Ēł¼Ļ│╝ĒĢśņ¦Ć ļ¬╗ĒĢśļŖö ĒĢ£Ļ│äĻ░Ć ņ׳ļŗż[14]. ĻĘĖļ¤¼ļéś ļćīņ▓ÖņłśņĢĪņŚÉ ņ¦üņĀæ ņĢĮļ¼╝ņØä ņŻ╝ņ×ģĒĢśļŖö ļō▒ ņ╣©ņŖĄņĀüņØĖ ļ░®ļ▓ĢņØĆ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņØś ļ¦īņä▒ņĀü ņ╣śļŻīļź╝ Ļ│ĀļĀżĒĢśņśĆņØä ļĢī ņĢłņĀäņä▒ ļ░Å ņ×äņāü ņĀüņÜ®ņŚÉ ņĀ£ĒĢ£ņØ┤ ļÉśĻĖ░ ļĢīļ¼ĖņŚÉ ņØ┤ļź╝ ĻĘ╣ļ│ĄĒĢśĻĖ░ ņ£äĒĢ┤ ĒŖ╣ņĀĢ ļŗ©ņØ╝Ēü┤ļĪĀĒĢŁņ▓┤ļź╝ ļČĆņ░®ĒĢ┤ BBBļź╝ ĒåĄĻ│╝ņŗ£ĒéżļŖö ņØ╝ļ¬ģ ĒŖĖļĪ£ņØ┤ļ¬®ļ¦ł(Trojan horse) ĻĖ░ļ▓ĢņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ļÅä ĒÖ£ļ░£Ē׳ ņØ┤ļżäņ¦ĆĻ│Ā ņ׳ļŗż[2,14]. ļśÉ TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņØś ļ¦Éņ┤ł Ēł¼ņŚ¼ļĪ£ļÅä ņżæņČöņŗĀĻ▓ĮĻ│äņØś TNF-╬▒ ņłśņżĆņØä ņĀĢņāüĒÖöņŗ£Ēé¼ ņłś ņ׳ņØīņØ┤ ĒÖĢņØĖļÉ£ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ ļ¦Éņ┤ł Ēł¼ņĢĮņØä ĒåĄĒĢ£ ņ╣śļŻī ņĀäļץļÅä ņłśļ”Į Ļ░ĆļŖźĒĢ©ņØä ņŗ£ņé¼ĒĢ£ļŗż[15].
ņØ┤ ņŚ░ĻĄ¼ļŖö ņØ┤ļ»Ė ņ×äņāüņŚÉņä£ ļäÉļ”¼ ĒÖ£ņÜ®ļÉśĻ│Ā ņ׳ļŖö TNF-╬▒ ņ¢ĄņĀ£ņĀ£ņØĖ adalimumabņØ┤ ņĢīņĖĀĒĢśņØ┤ļ©Ėļ│æņØś ļīĆĒæ£ņĀü ļ│æļ”¼ĻĖ░ņĀäņ£╝ļĪ£ ņĢīļĀżņ¦ä A╬▓1-42ņØś ņŗĀĻ▓Į ļÅģņä▒ņŚÉ ļīĆĒĢ£ ļ│┤ĒśĖ ĒÜ©Ļ│╝, ņ”ē NeuNĻ│╝ Bcl2 ļŗ©ļ░▒ņ¦ł ļ░£ĒśäņØś ņ”ØĻ░ĆņÖĆ tau ļŗ©ļ░▒ Ļ│╝ņØĖņé░ĒÖöņØś ņ¢ĄņĀ£ĒĢśļŖö Ļ▓āņØä ĒÖĢņØĖĒĢśņśĆņ£╝ļ»ĆļĪ£, Ē¢źĒøä ņĢīņĖĀĒĢśņØ┤ļ©Ė ņ╣śļ¦ż ĒÖśņ×ÉņŚÉ ņĀüņÜ®ĒĢśļŖö ĒøäņåŹ ņŚ░ĻĄ¼ņØś ņ¦äĒ¢ēņØ┤ ņÜ®ņØ┤ĒĢĀ Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż. ļŗżļ¦ī ņäĖĒż ņłśņżĆņØś ņŚ░ĻĄ¼ņØ┤ļ»ĆļĪ£ ļÅÖļ¼╝ņŗżĒŚśņØä ĒåĄĒĢ┤ ņØĖņ¦ĆĻĖ░ļŖźņØś Ļ░£ņäĀĻ│╝ BBBņØś ĒåĄĻ│╝ ļō▒ņŚÉ ļīĆĒĢ£ ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśĻ▓Āļŗż.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print